NGS 注意事项:覆盖率、读取长度、多路复用
规划下一代测序 (NGS) 实验时需要考虑几个因素。这些考虑因素包括:测序覆盖的深度、测序读取的长度、是否进行单端或双端测序以及多路复用选项。由于测序成本将根据这些决定而有所不同,因此规划实验以获得回答实验问题所需的适当数量的测序数据非常重要。这篇文章将帮助您解决这些考虑因素,特别是关于免疫组库测序项目。有关更多背景信息,请参阅我们的NGS 简介文章。
测序覆盖率
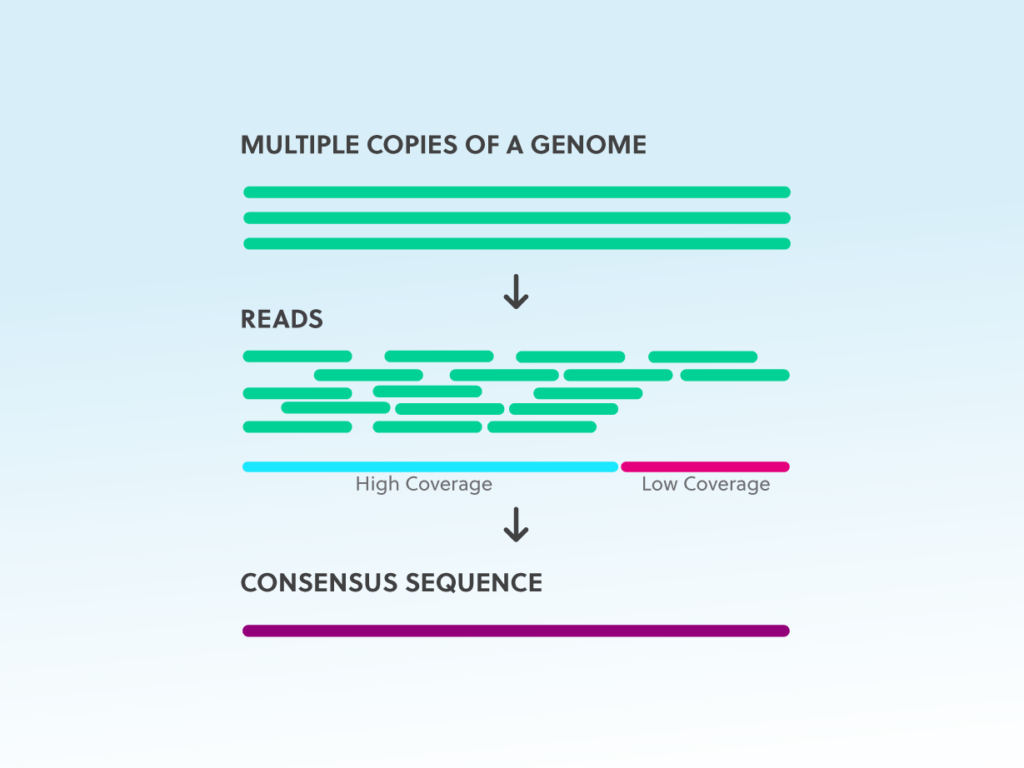
序列覆盖率(深度)描述的是目标转录本或基因组中特定位置上与已知参考序列对齐的平均读取次数。由于测序容易出错,因此使用更高的覆盖率来提高对所读取碱基的置信度。采用这种方法的前提是测序错误是随机的。因此,如果每个核苷酸被测序多次,则大多数读取(共识)共享的碱基读取将反映正确的核苷酸。

在 NGS 中,序列读取需要多次覆盖每个碱基以增加置信度;当单个测序读取错误的数量超过正确读取数量时,它们在统计上是无关紧要的。样本内的覆盖率是可变的,对于典型的人类遗传和癌症应用,典型覆盖范围分别为 30 次或更少到 >1000 次读取。
有必要确定您的应用所需的测序覆盖范围,以最大限度地降低错误结果的概率。例如,如果您想捕获转录组中的稀有 RNA 序列,则需要进行更深层次的测序以检测低丰度变异。
免疫组库测序不同于其他测序应用,因为每个细胞都有可能包含其自己独特的 VDJ 序列。因此,每个细胞都代表从头重排。在靶向 VDJ 测序应用期间,VDJ 重排,特别是 CDR3 区域,被直接捕获和测序,因此不需要对短读片段进行额外的生物信息学组装(除了可能的读段拼接,在下面的“测序读段长度”部分中讨论)。在免疫组库测序应用中,测序覆盖率是根据样本的起始细胞数和样本多样性估计值(如果已知)确定的。需要为每个样本分配足够的读段以覆盖基于输入细胞计数的潜在多样性。
人类白细胞的平均数量为 4,000 至 11,000 个细胞/µL。细胞在所有处理步骤中都会丢失,因此在提取 RNA 之前对样本的最终细胞数量进行估算非常重要。我们通常建议为样本中每个细胞数分配至少 5-10 倍的读取次数。因此,对于含有 100,000 个细胞的样本,应分配至少 500,000 个读取次数。这意味着对于细胞数量通常较低的样本,有机会在每个通道(或流动池)中汇集更多样本,限制是目标链的可用分子 ID 或条形码的数量。如果样本或 RNA 源预计具有有限的库,则可能不需要进行非常深入的测序。例如,Jurkat 细胞系只有一个 TCR-beta 重排。在这种情况下,细胞计数在确定所需读取数时无关紧要,因为只需很少的读取即可获得序列。为此类样本分配过多的读取会不必要地浪费昂贵的流动槽空间。过度测序还会导致低频测序错误的累积。
测序读长
读长描述了产生的测序读长的平均长度(即测序的碱基对数),并且是特定于测序平台的。如果将读长组装成重建的 DNA 序列就像拼拼图一样,那么长读长就相当于更大的拼图块。对于全基因组测序或物种识别,较长的读长是更好的选择。较长的读长对于捕获插入和删除或对具有大量冗余的区域(例如包含转座子的区域)进行测序也是必不可少的。
短读取对于计数特定序列的丰度、识别其他保存良好的序列中的变体或分析特定转录本的表达的应用非常有效。
NGS 读取长度和覆盖率是两个可以控制的参数,需要在测序前预先确定。由于测序成本是按每个碱基对计算的,因此通常在测序深度和测序长度之间进行权衡。与短读取测序相比,执行较长读取的测序平台通常会提供较少的覆盖率、更高的错误率和更高的每碱基成本。
测序读取类型:单端读取与双端读取
术语“双端”读取是指在测序过程中读取同一 DNA 序列的正向和反向模板链。
由于两端之间的距离是已知的,因此可以使用此信息将读取映射到重复区域上。双端读取提高了检测测序读取相对位置以及识别基因插入、缺失、重复序列和其他重排的能力。由于它与免疫组库有关,双端读取还可用于扩展 VDJ 受体的覆盖范围。正向和反向读取可以从 V 和 C 方向进行测序,可以使用读取之间的一小块重叠区域将它们拼接在一起。该重叠区域可用于延长读取长度,以便可以从框架 1 内到 C 区域开头提供受体信息(有关更多信息,请参阅我们关于iRepertoire 引物系统的帖子和我们的生物信息学平台的技术说明)。
双端测序涉及两倍的测序量,因此成本更高,但其优势在于准确性更高,读取长度更长。因此,可以直接捕获整个 VDJ 区域,而无需使用任何复杂的下游基因组组装。单端读取以相同的成本提供更大的覆盖范围,非常适合那些不看重延长读取长度或可以通过其他方式(例如,独特分子索引)提高准确性的研究。对于 VDJ 分析,数据会在读取拼接过程中丢失;但是,双端读取数据具有可以作为单端或双端进行分析的优势。如果下游应用只需要独特 CDR3 区域的频率,那么单端数据足以进行频率计算和 CDR3 识别,而不会遭受数据丢失。但是,如果需要扩展信息以识别 CDR1 和 CDR2 区域,则可以将相同的数据作为双端和读取拼接进行分析。
多路复用文库
一次测序运行产生的数据量通常比给定项目所需的数据量还要多。为了节省成本,可以通过合并或多路复用将多个样本合并到一次测序运行中。为了从合并的测序运行中重新提取单个样本,分子条形码通过用于在文库制备过程中扩增序列的引物附加。条形码使数十个甚至数百个不同的样本能够多路复用到一次测序运行中。当针对多个不同样本中的基因组特定区域时,多路复用是理想的选择。
数据分析软件通过自动对具有不同条形码的读数进行分类,帮助在运行后对测序数据进行解复用。将样本复用到一次运行中意味着您将获得每个样本的数据较少。每个样本的覆盖率大致等于总覆盖率除以样本数量。有关更多信息,请参阅我们关于文库池的帖子。
文库制备挑战
为了制备用于 NGS 的 DNA,可以将测序接头连接到 DNA 片段的末端或通过 PCR 添加到 DNA 片段的末端。文库制备工作流程可能包括几轮 PCR 扩增。由于 PCR 的指数性质,扩增模板混合物具有挑战性。扩增过程中非特异性或副产物的形成会降低所需信号并给结果数据带来不良噪音。例如,小片段优先被扩增;这意味着小扩增子可以快速占据文库,而大扩增子则被排除在外。在文库制备过程中排除(通过实验设计)或去除(通过纯化步骤)不需要的材料可减少 PCR 扩增产生的噪音;这可能包括不需要的或人造的小扩增子,例如引物二聚体。这会增加所需产品的信号,最终获得更好的数据。
要了解 iRepertoire 的测序技术如何排除不需要的扩增子(包括引物二聚体),请参阅我们的扩增技术帖子。